-
GBrowse
Refer to this page for general use of GBrowse.
Data search
VigGS offers simple keyword search and sequence similarity searches using BLAST or BLAT.
Keyword search
There are two search boxes in VigGS; one is on the top page, and the other is on the menu bar.


Just putting any words in the search box and pressing the Enter key will execute searching. Search results are shown in a window like this:
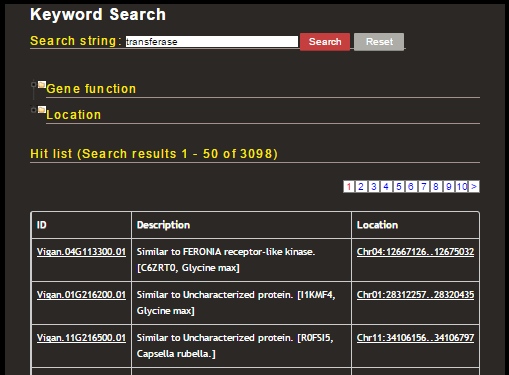
In this case, 3098 genes were hit with a keyword 'transferase.' The 'ID' column of the gene list links to the detailed annotation and 'Location' column links to the GBrowse. You can further filter the genes by putting another keyword or by selecting any functional domain.
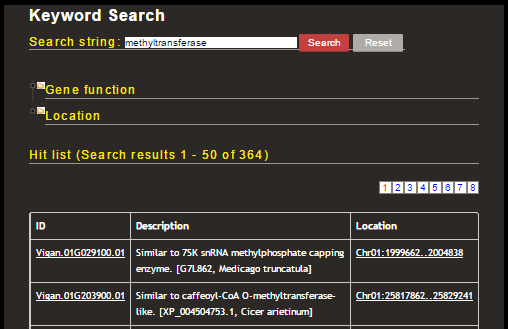
Now the 3098 genes have been narrowed down to 364 by putting 'methyltransferase.' Note that the text box shown on top of the result page is only for narrowing down the genes. If you want to start over the search, put the new keyword in either of the two search boxes as shown above. In order to select genes with any functional domain, first click the folder icon shown at 'Gene function' and expand the list of functional domains.

Then select any Gene Ontology (GO) or InterPro domain and the list of genes will be updated. Likewise, you can select genes on a specific chromosome or scaffold in the 'Location' section. In order to select genes on a specific region, specify the region in the text box in the form of '<sequence id>:<start position>..<end position>'.
BLAST/BLAT search
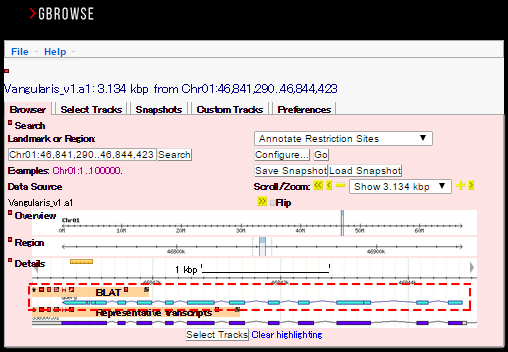
You can search any DNA sequences by BLAST (blastn, blastx, or tblastx) or BLAT, or protein sequences by BLAST (blastp or tblastn) against the azuki bean genome or gene sequences. Query sequences can be in multi-FASTA format. If you select genome sequence as the target database, you will see the GBrowse icon in the result page as shown below (in the red dashed line).


You can refer to the structure of the query sequence on the genome with any other tracks in parallel.
-
Variant information
Currently variant information including SNPs and small indels detected between azuki bean and its close relative, V. nepalensis, are implemented in VigGS. There are three ways to refer to the variat information:
By selecting any variant on GBrowse
If you select any variant shown on GBrowse, detailed information about the varinat will be shown in the GBrowse details page.
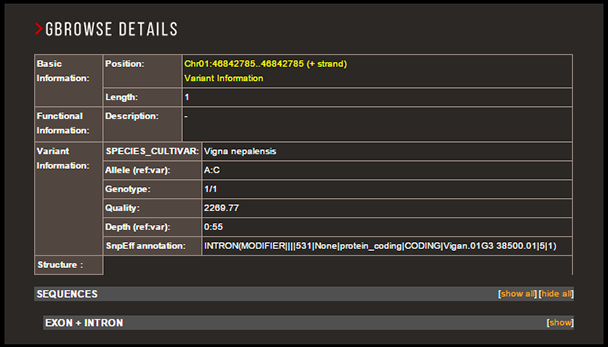
From the GBrowse details page
In the GBrowse details page of each azuki bean gene, there is a link to the variant information.

This link will show you a list of variants found within the target gene (see below).
By selecting a genomic region on GBrowse
If you want to see the variant information within a genomic region of your interest, select any region on GBrowse and you will see a link to the variant information in a pop-up window as shown below (in the red dashed line). This link will show you a list of variants found within the region (see below).
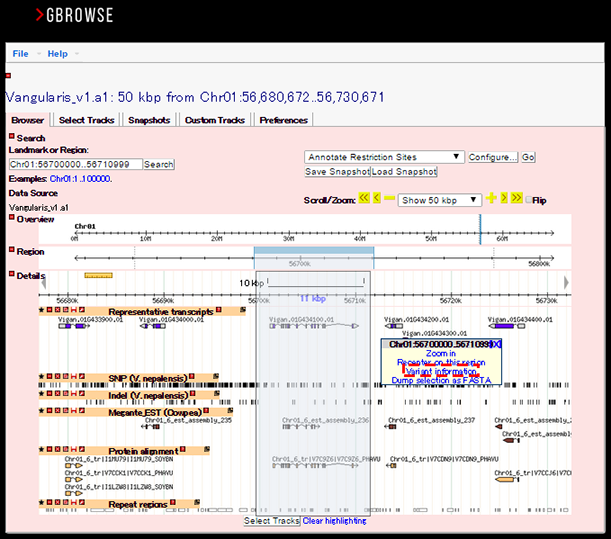
List of variants found within a selected gene or genomic region are shown like this:
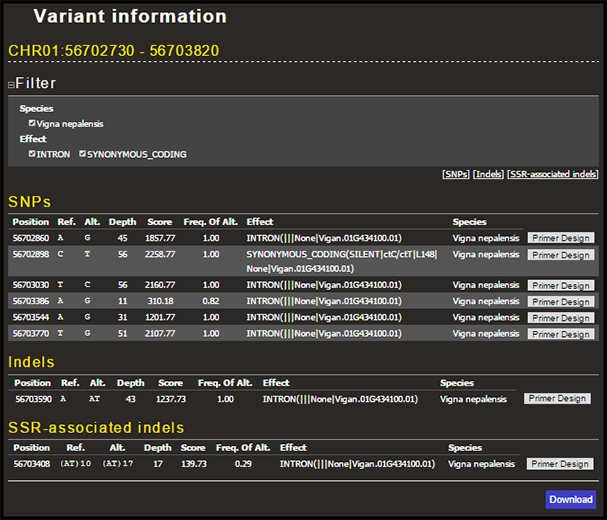
Variants are shown by type: SNPs, indels, and Simple Sequence Repeat (SSR)-associated indels. You can filter the variants by type of effect assigned by SnpEff. The variant information can be downloaded in VCF format by clicking 'Download' button at the bottom right. The 'Primer Design' button shown in each line links to another page where you can design primer pairs by Primer3 targeting the selected variant.
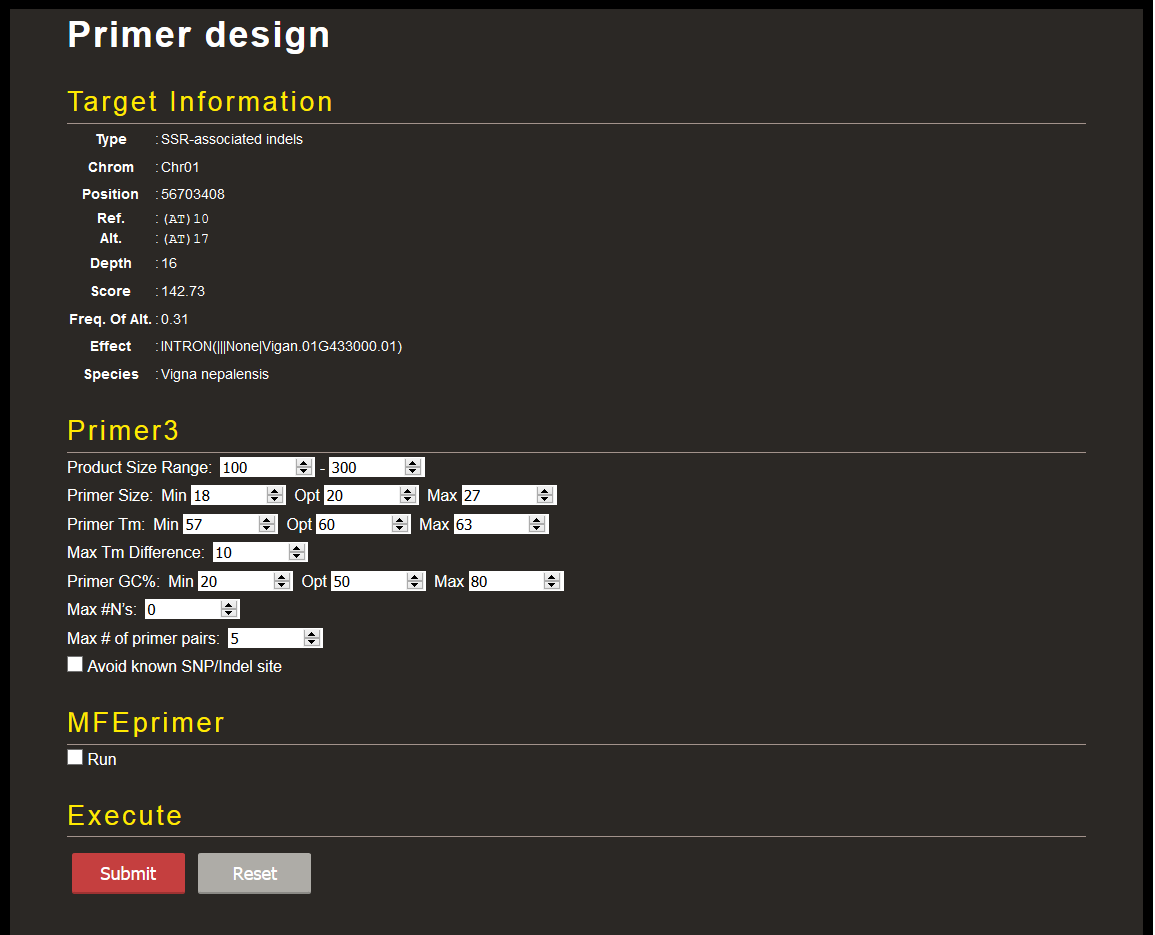
You can also check the specificity of the picked primer sequences by MFEprimer against the genome sequence. To do that, check the box at 'MFEprimer' section, set options, and click 'Submit' button.
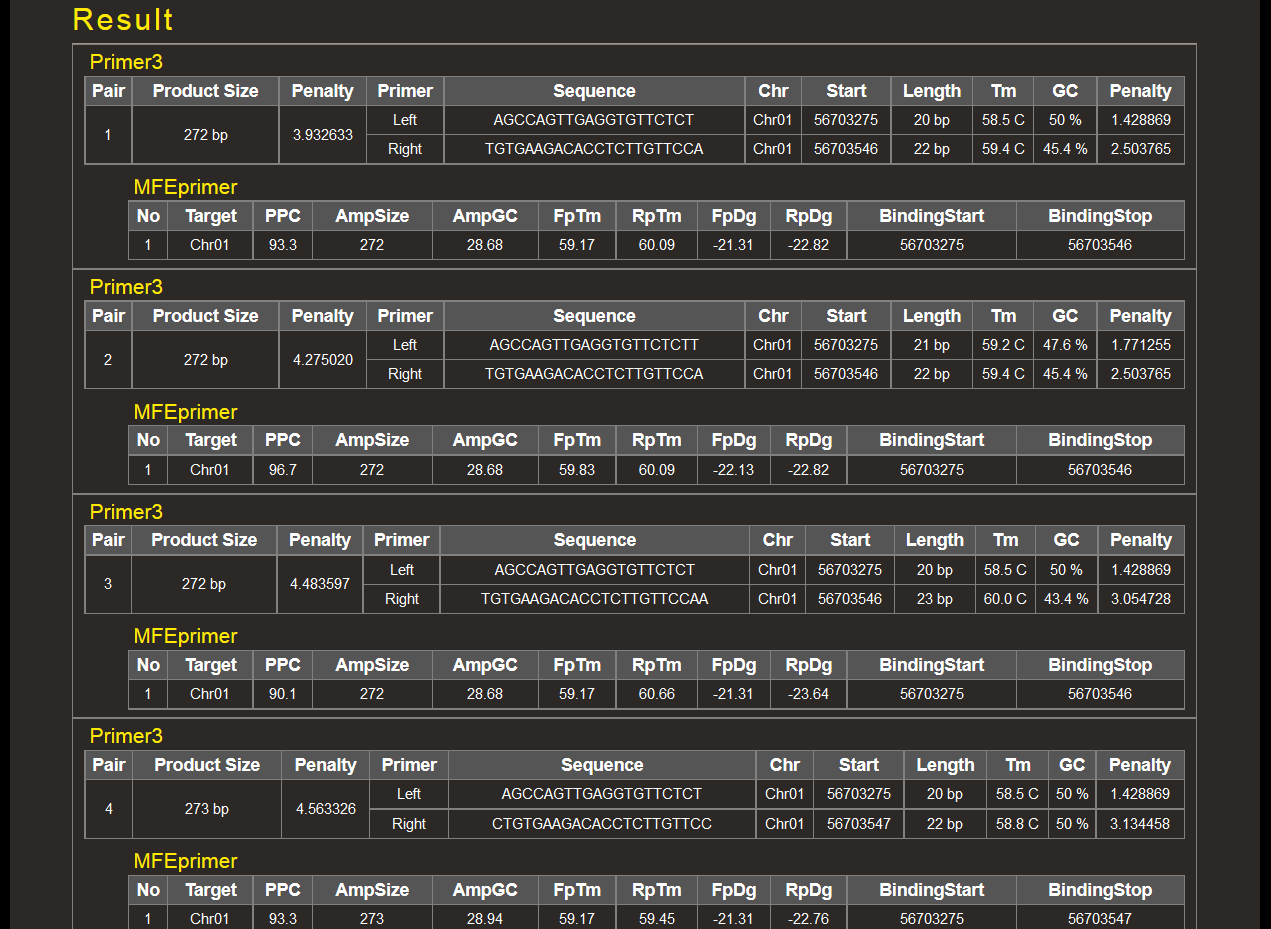
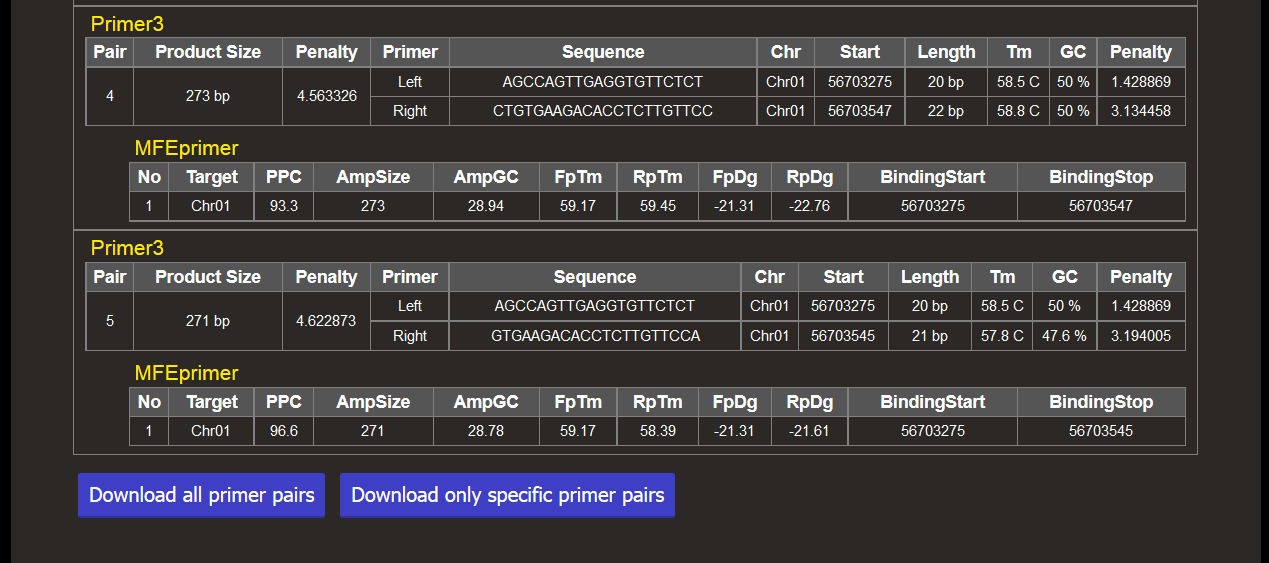
Results are shown as below. If any pair of primer sequences is mapped to multiple locations, the primer pair is dealt with a non-specific pair.
You can download either all primer pairs or only specific primer pairs in a tab-delimited text file.
-
Designing primer pairs directly from GBrowse
You can design primer pairs targeting any sites and any regions on the genome. To do that, select any region on GBrowse and you will see "Primer design" menu in a pop-up window as shown below. This link will take you to another page where you can design primer pairs as shown above.

-
Multiple alignment
In the GBrowse details page of each azuki bean gene, there is a link to another page where you can construct multiple-gene alignment among orthologous genes in six dicot species as well as azuki bean paralogous genes.

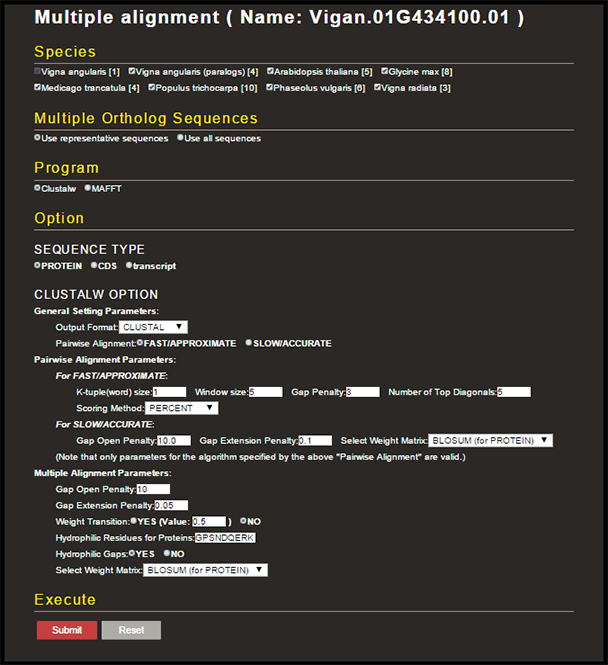
Orthologous genes have been predicted among Arabidopsis thaliana, Glycine max, Medicago truncatula, Phaseolus vulgaris, Populus trichocarpa, and Vigna radiata. Numbers in the parentheses represent the number of orthologous genes in each species. If you select 'Use representative sequences' in the 'Multiple Ortholog Sequences' section, only the longest gene in each species will be used in the alignment process (default). You can align protein, CDS, or transcript sequences (default: protein) by Clustalw or MAFFT (default: Clustalw). Constructed multiple-gene alignment as well as the query sequences can be downloaded.

-
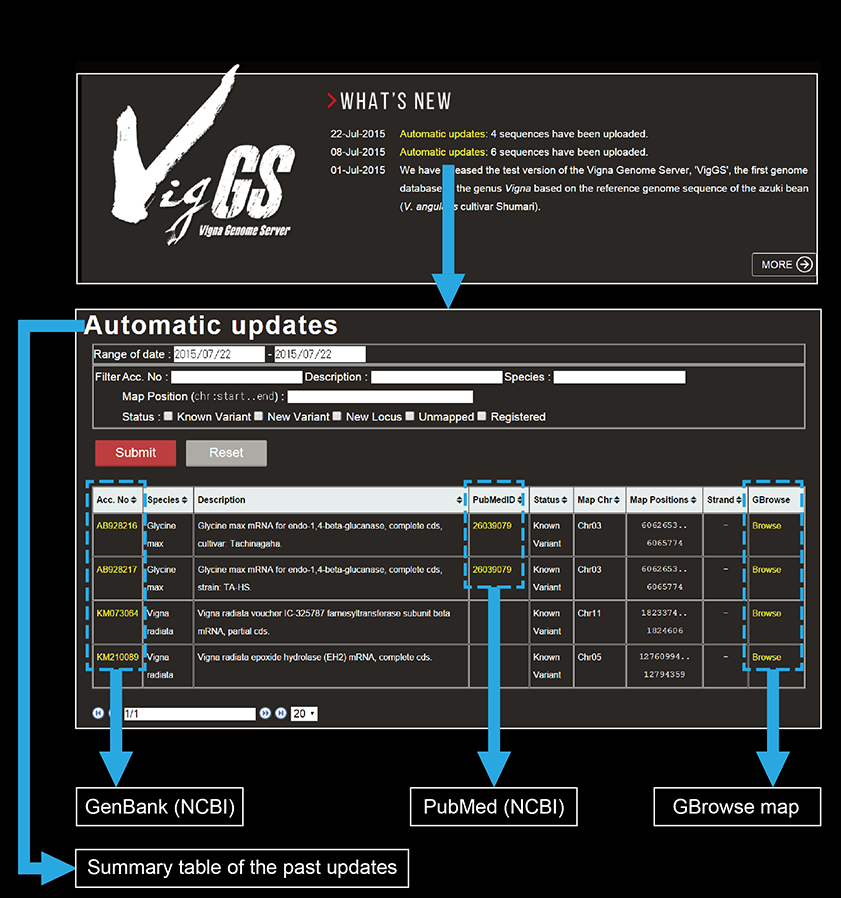
Automatic updates
Currently VigGS downloads mRNA sequences of G. max, P. vulgaris, M. truncatula, and any Vigna species once a week from NCBI GenBank. Number of the obtained sequences is reported in the "What's new" window on the top page, which links to a table showing the accession numbers, species names, descriptions, PubMed IDs, mapping results, and links to GBrowse map for the submitted sequences. Title of the table, 'Automatic updates', links to the summary table of the past updates. Obtained sequences are mapped to the azuki bean genome by BLAT or Exonerate (if CDS are indicated in the sequence record). Mapped sequences are uploaded to GBrowse and displayed on the “Automatic Updates” track, with links to the original GenBank and PubMed records in the GBrowse details page. In addition, exon/intron structures of the mapped sequences are compared with annotated azuki bean genes. If all the intron positions are consistent with those of any of the annotated genes, such sequences are reported as “Known variant” or otherwise reported as “New variant” if at least one of the introns is unique. Mapped sequences without any overlapping annotated genes are classified into the category “New Locus”. Users can filter sequences by accession number, description, species name, map position, and status.